Software
Tools for Environmental DNA Analysis and Data Interpretation
eDNA Viewer
The eDNA Viewer is an interactive platform for exploring biodiversity data generated through environmental DNA (eDNA) metabarcoding, developed in collaboration with eDNA Frontiers. The tool allows users to visualise species detections, examine taxonomic composition, and investigate patterns across space, time, and habitat using curated and standardised datasets from marine, freshwater, and terrestrial ecosystems across Australia and beyond.
The platform features an interactive map for spatial exploration, sample and project browsing, taxonomic trees, diversity and abundance summaries, and multivariate analysis tools. Users can filter by location, species, sample type, assay, and collection timepoints, and export selected datasets for further analysis. Public datasets are immediately accessible, while registered users can log in to access additional project-specific data according to their permissions.
The eDNA Viewer can be found at: https://viewer.ednafrontiers.com/#!/map
FAQs
What data is in the Viewer?
There are two levels of access to this app – public and private.
Public projects are openly accessible to anyone visiting the app, offering basic functionality such as taxa searches without requiring a login via the sample page.
Private projects have been processed by eDNA Frontiers but not yet released to the public by their owners. Logged-in users can access their private data along with expanded functionality via the project page, such as graph and tree building.
Can I make my data public?
We encourage all of our users to make their data publicly accessible. If you have completed a project with us and would like to release your data onto our Public platform, please email us at ednafrontiers@curtin.edu.au and let us know!
How do I access my data?
If you have completed a project with us and would like an eDNA Viewer account to access your data online, please email us at ednafrontiers@curtin.edu.au
Can I download data?
All of our tables and graphs can be downloaded from the Viewer. You can even choose whether to download the filtering parameters used to generate the data, so you don’t forget!
Can I share data?
Any user with a login can give permission for one (or all) of their projects to be shared with someone else. Just email us at ednafrontiers@curtin.edu.au and let us know which projects you would like to assign to another user.
Seqema
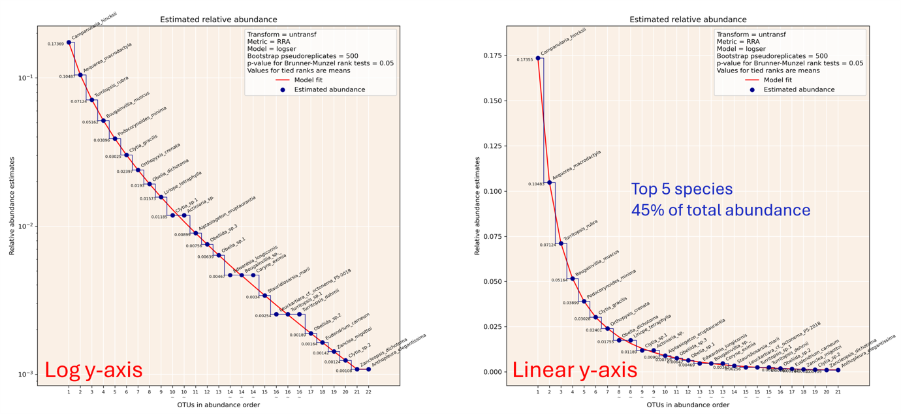
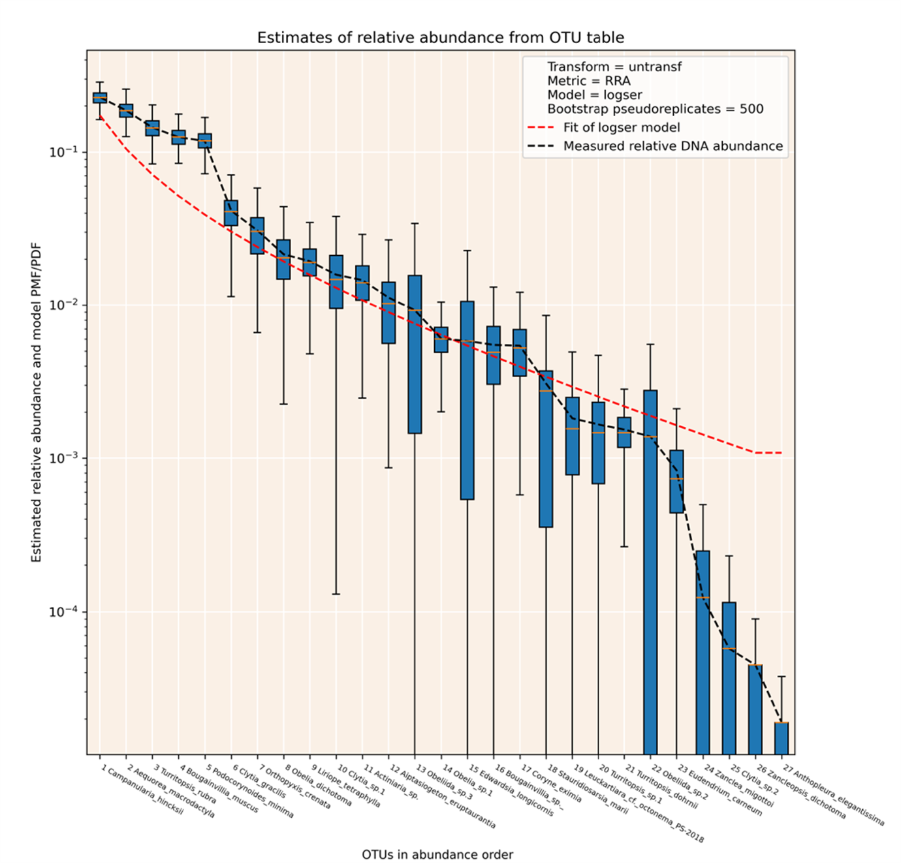
eDNA metabarcoding produces results with a quasi-quantitative relationship between molecular abundance and the biomass of organisms represented by DNA barcodes. The relationship is imperfect for two types of reasons: stochastic factors and consistent biases.